 hotline服务热线:010-61006450
hotline服务热线:010-61006450
文献解读 | 用生理学模型评价食物对克拉霉素速释片口服吸收的影响-九游会电竞
生理药代动力学(pbpk)等模型的建立有助于预测体外特征对药物吸收的影响。本文以《evaluation of food effect on the oral absorption of clarithromycin from immediate release tablet using physiological modelling》文献为依据,通过生理吸收模型预测克拉霉素速释片在体内吸收的曲线及分布,同时采用机械吸收模型建立体内外相关性(ivivr),详细解读ph、胆汁分泌和食物对克拉霉素速释片口服吸收的影响。
体外检测克拉霉素在各介质中的溶解度、片剂崩解和溶出曲线,以及高脂餐匀浆后的流变。
✎ 降解:克拉霉素在ph3-8水溶液中较为稳定,在胃ph(1-2)下会迅速降解。室温下,在0.1n hcl介质中1h内便会降解50%。
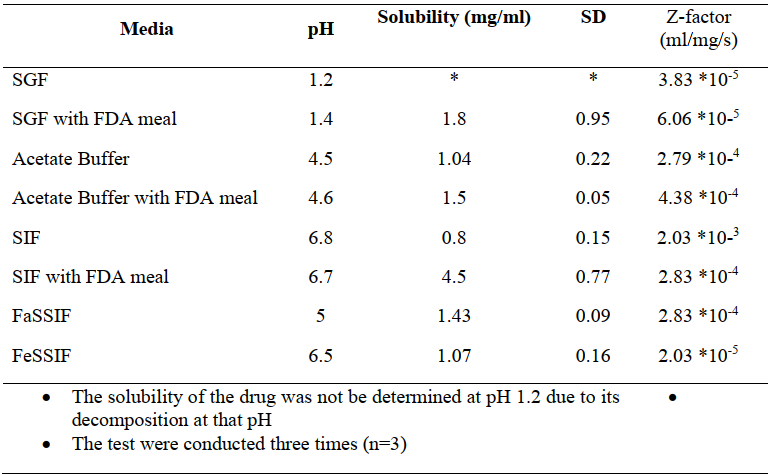
✎ 溶解度:克拉霉素的溶解度无ph依赖性,由于克拉霉素在酸性条件下会快速降解,无法准确测得ph1.2的溶解度。检测数据表明,由于高脂餐中的高脂肪含量可显著提高亲脂性药物(bcsii类)的溶解度,食物会促进克拉霉素的溶解。
表1 克拉霉素在不同介质中的溶解度和转换因子
✎ 流变:稀释后高脂餐的流变性结果表明,食物的流变性具有假塑性,随剪切速率增加粘度降低。高脂餐的粘度具有ph依赖性,粘度与ph呈正相关。同时,在sif和醋酸盐介质中,高脂餐的流变曲线具有叠加性。
✎ 崩解:介质ph值对克拉霉素片的崩解影响显著,由于片剂表面会生成凝胶,克拉霉素片在酸性介质中崩解时间较碱性介质中偏长。在不同介质中加入食物,也会延迟药物的崩解,这是由于加入食物后介质粘度变高。
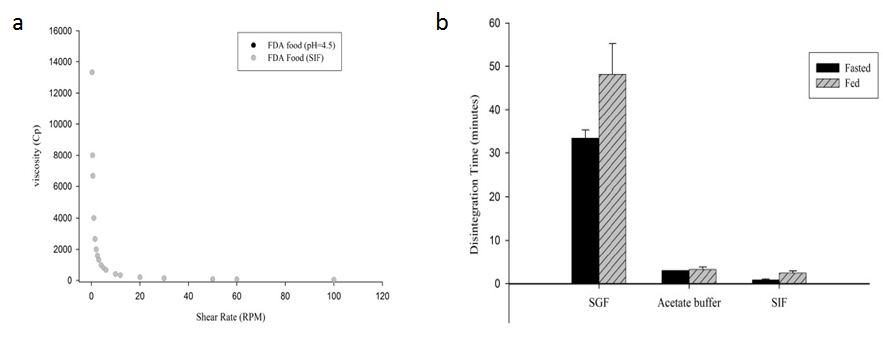
图1 a:标准高脂餐(醋酸盐和磷酸盐缓冲液)在不同剪切速率下的粘度;b:克拉霉素在不同介质中的平均崩解时间
✎ 体外药物释放影响因素:
-
介质ph:克拉霉素片在ph4.5和ph6.8介质中溶出较为完全,但在ph1.2介质中溶出终点仅为15%。这是因为克拉霉素在酸性介质中发生降解,且片剂表面形成凝胶而减慢了克拉霉素片的崩解。
-
食物:食物对药物的体外释放影响显著,但各ph中食物对药物释放的影响不尽相同。在人工胃液(sgf)中食物会促进克拉霉素释放,减缓降解速率。然而,在ph4.5和ph6.8介质中,片剂崩解缓慢,食物降低了克拉霉素的吸收。
-
胆酸盐:测定克拉霉素在生物相关介质和常规介质中释放。克拉霉素在禁食状态肠液和禁食状态胃液介质中释放均比常规介质中偏低。在禁食状态肠液(ph6.5)和磷酸盐缓冲液(ph6.8)介质中120min分别释放60%、88%,两个介质ph值仅差0.3,但释放百分比相差28%。克拉霉素在禁食状态胃液(ph=5)释放非常低,120min仅释放10%。对比空腹和餐后状态各介质中的药物释放,f1和f2均小于50,二者不等效。
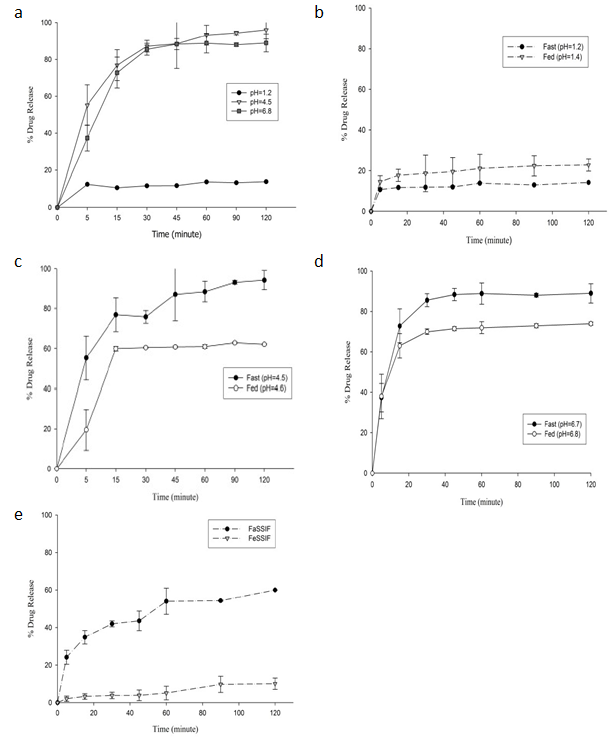
图2 a:克拉霉素速释片(500 mg)在各介质中的溶出曲线;b:克拉霉素在胃液(空腹和餐后)介质中溶出曲线;c:克拉霉素在醋酸盐(空腹和餐后)介质中溶出曲线;d:克拉霉素在磷酸盐缓冲液(空腹和餐后)介质中溶出曲线;e:克拉霉素在生物相关介质中的体外溶出曲线(禁食状态肠液与禁食状态胃液)
01、药物吸收建模:
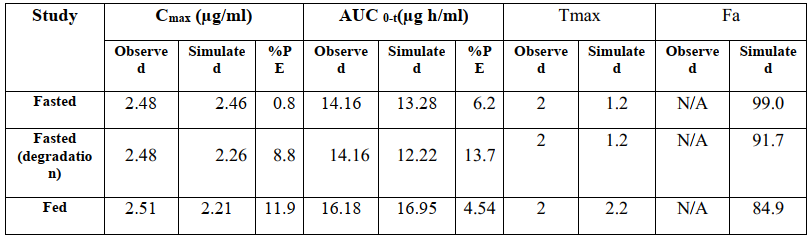
使用gastroplus软件pbpk模型对口服给药的克拉霉素500mg速释片进行建模,部分建模参数如表2,其他参数为软件默认。模型预测参数与观测值详见表3,经比较,预测值与观测值误差不超过10%,表明此次建模具有较好的预测性。由于食物减慢了胃排空速率,餐后给药tmax延长。同时,模拟结果表明食物对cmax和auc无显著性影响。
表2 建模输入参数
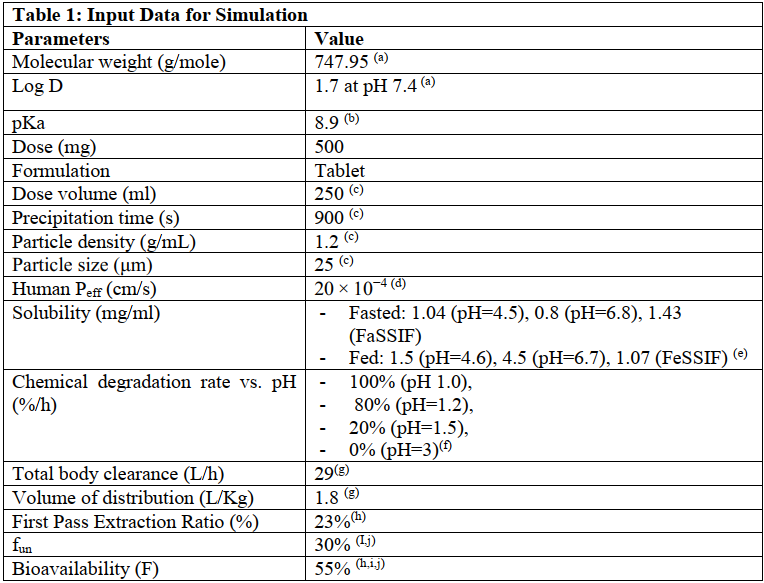
表3 pk参数的建模预测值与观测值
研究药物的酸降解对克拉霉素口服生物利用度的影响发现,考虑酸降解时cmax和auc减小。克拉霉素在胃肠道吸收快速,且模型预测空腹条件下克拉霉素吸收分数为91.6%,餐后条件下吸收100%。考虑原因为克拉霉素在胃ph环境中不稳定,降低了药物的吸收。空腹状态下克拉霉素67.1%吸收分布在十二指肠,27.1%的剂量在空肠。餐后状态下,克拉霉素吸收完全,由于胆汁再摄取增强,73.7%剂量比例吸收分布在十二指肠,22.1%分布在空肠。
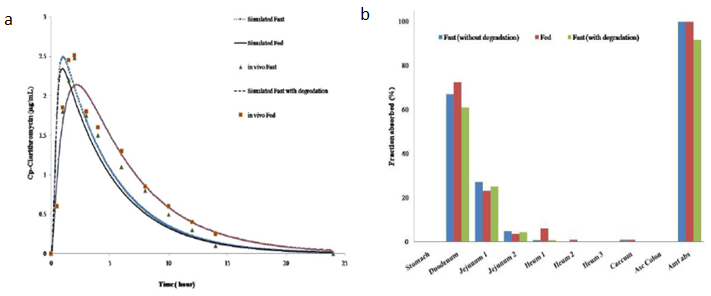
图3 a:空腹和餐后试验下克拉霉素模拟和观测血浆浓度;b:空腹和餐后试验下克拉霉素的吸收分布
✎ 敏感性分析:选择参数溶解性、渗透性、胃排空速率、胃肠ph、胃排空时间、总清除率、肝肠首过效应(fpe)对空腹及餐后试验条件下克拉霉素的吸收进行敏感性分析。根据结果,清除率、肝脏首过效应和肠道首过效应对克拉霉素的cmax及auc具有显著影响。然而,溶解性、渗透性、胃肠ph对克拉霉素的吸收无影响。餐后试验条件下,cmax受胃排空速率影响,cmax随胃滞留时间延长而降低。经敏感性分析发现,广泛的肝脏代谢和肠道首过效应可能会导致pk参数大幅度的下降,给药后肝血流量增加也会降低肝提取率并增大药物生物利用度。
02、虚拟be模拟
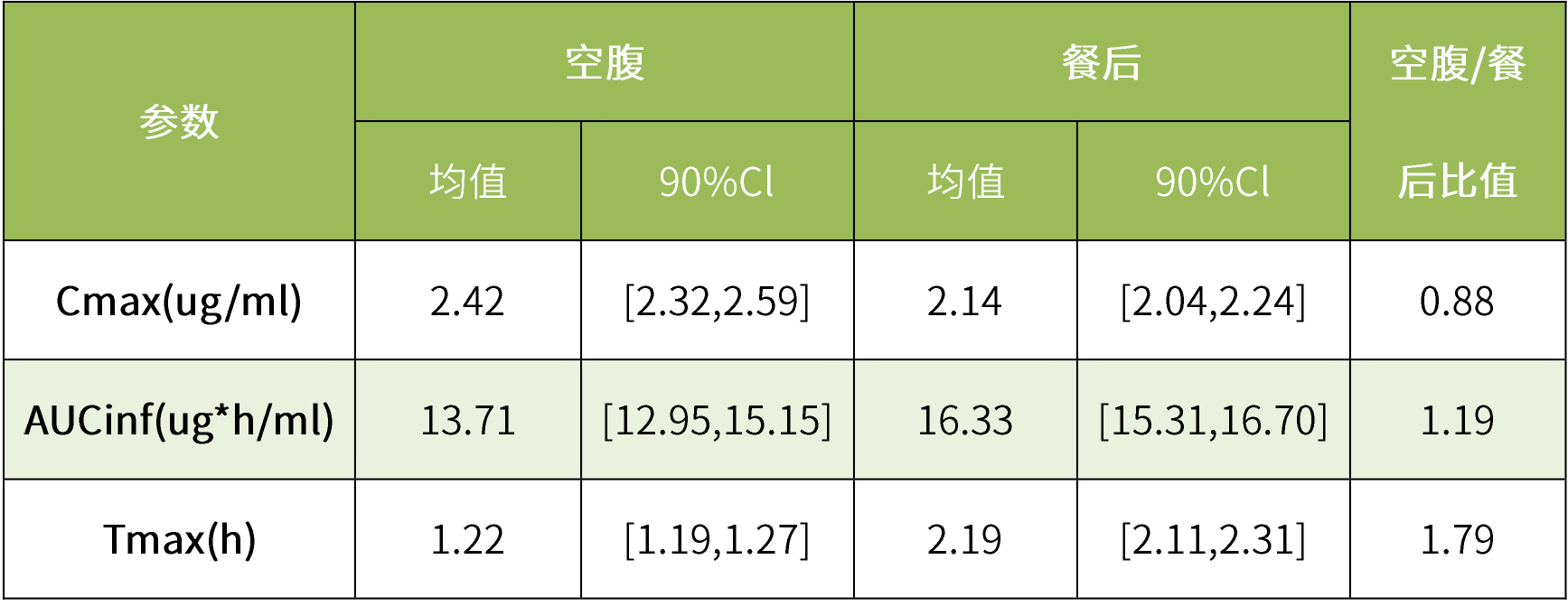
虚拟be模拟结果表明,食物对克拉霉素生物利用度无显著影响。克拉霉素片在空腹和餐后状态下的平均血浆浓度-时间曲线均落在90%置信区间内。虚拟人群试验表明,食物不会显著改变克拉霉素片的cmax和auc,空腹与餐后的cmax和auc的90%置信区间均在生物等效性限值内。
表4 克拉霉素的虚拟食物影响研究
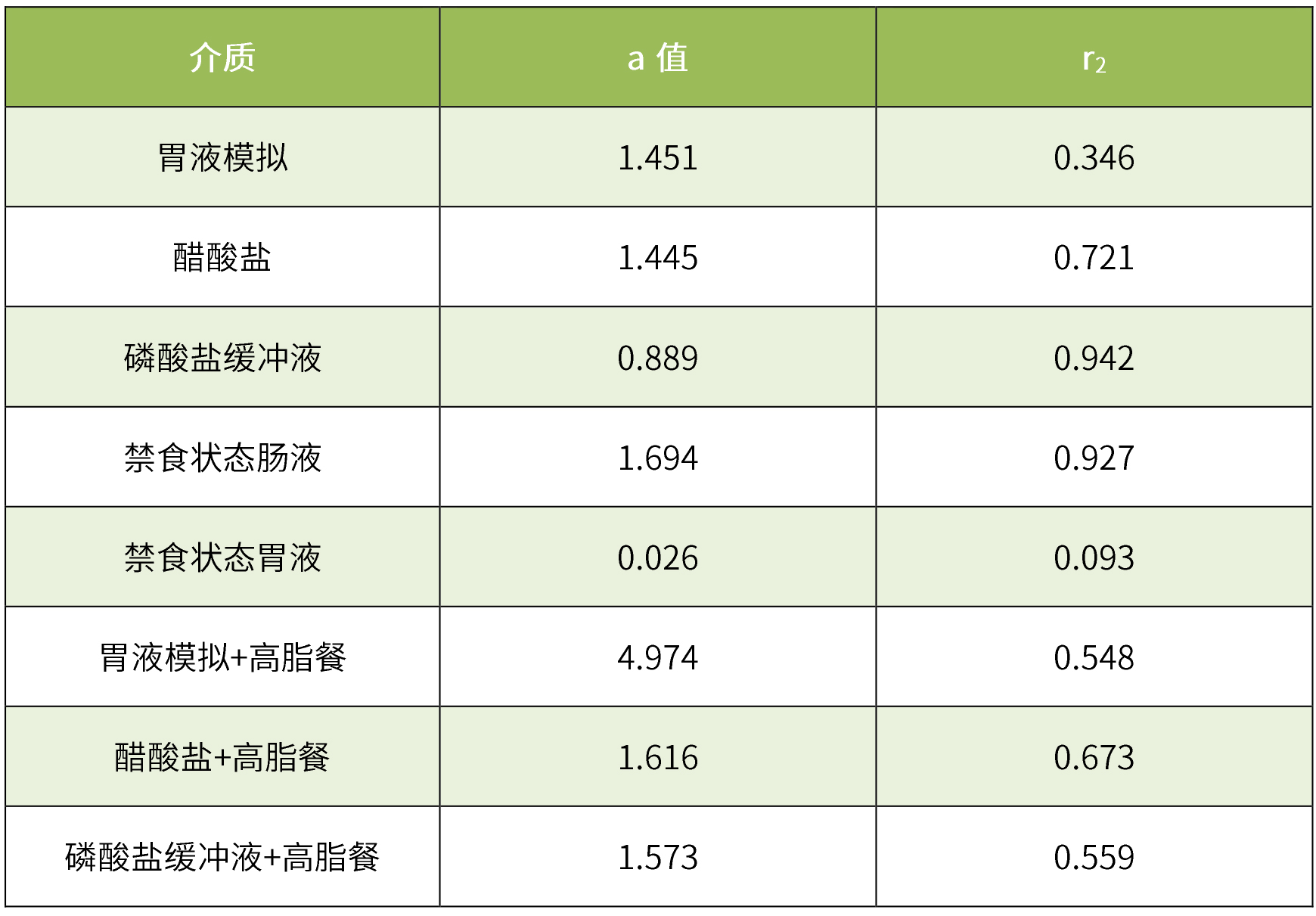
采用去卷积方法,利用机械吸收模型,将空腹和餐后状态下的体外溶出数据和相应的体内血浆浓度关联起来,建立体内外相关性。在磷酸盐缓冲液、醋酸盐和禁食状态肠液介质中成功建立了ivivr关系,相关系数(r2)值接近于1。但是,在hcl介质中体内外相关性较差(r2=0.3),餐后状态下不同介质的体外溶出与体内相关性均不佳。
表5 ivivr统计参数
模型验证
我司拥有国内首个体内外桥接(ivivr)研发平台,不仅拥有gastroplus软件,而且完成了克拉霉素片仿制药的申报。小编这里运用gastroplus软件的pbpk模型对克拉霉素片进行模型验证。将我司获得的餐后状态下的体内pk数据输入该模型中,模拟结果见表6。根据结果可知,我司取得的餐后pk数据与模型观测值拟合度极高,误差小于10%,二者血浆浓度药时曲线也几乎完全吻合,标明该模型具有良好的预测性。
表6 研发中克拉霉素片与模型拟合结果
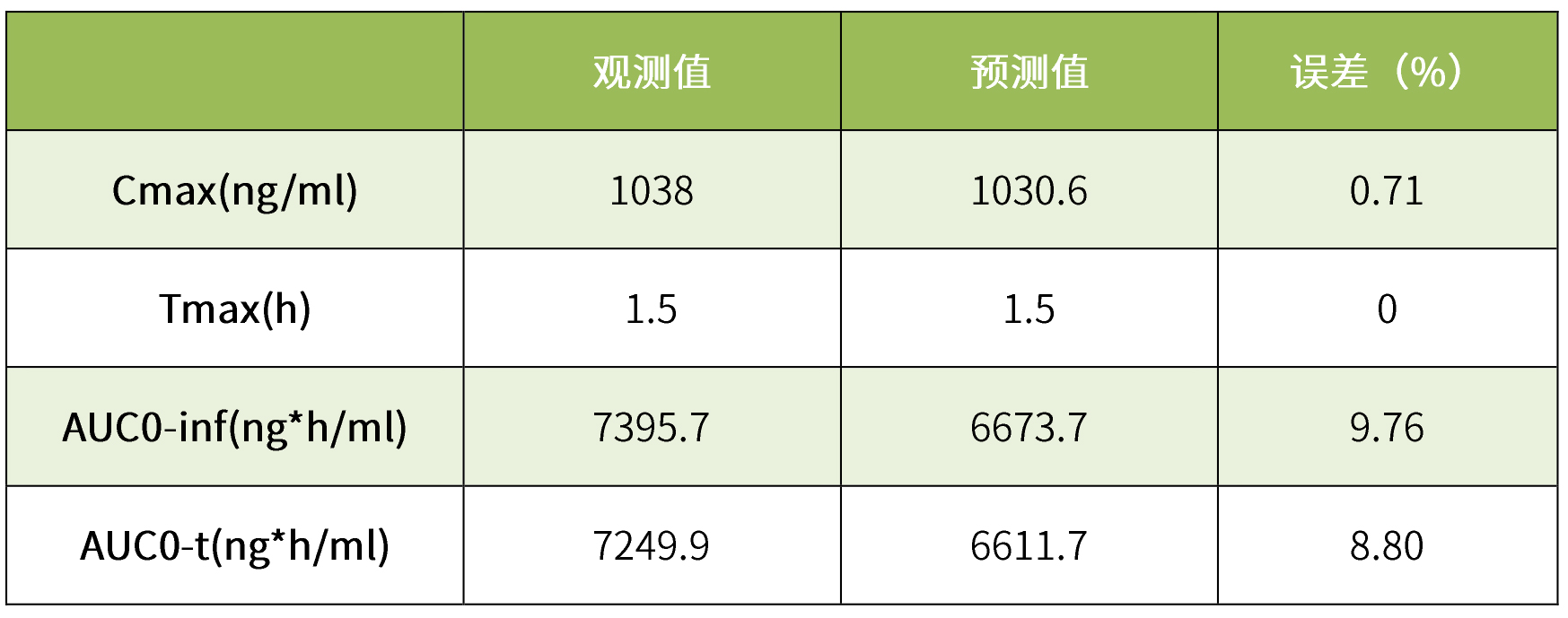
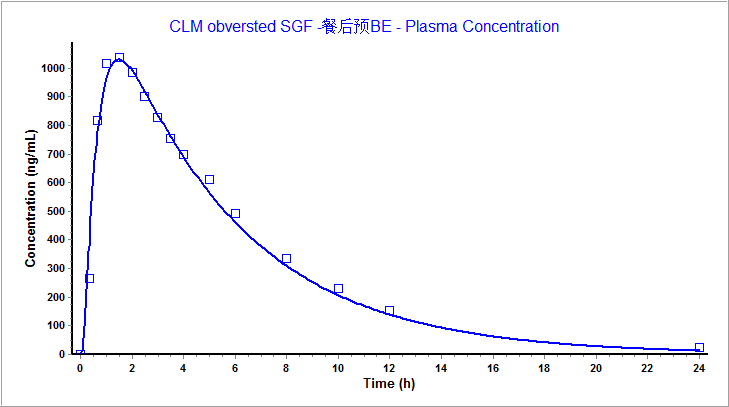
图5 克拉霉素片在餐后状态下与模型血浆浓度药时曲线拟合图
四、 讨论
本研究采用gastroplus建模和体外试验,研究了食物对克拉霉素口服生物利用度的影响机制,ph值、胆酸盐和食物对克拉霉素的口服吸收均具有显著影响。
-
酸性条件下,克拉霉素与盐酸反应形成凝胶,使其崩解时间变长。该凝胶层在中性介质中溶解,可加快药物崩解,促进药物释放。
-
在生理相关介质内,胆酸盐存在时克拉霉素的释放较常规介质中低很多,是因为大环内酯类抗生素与胆汁盐发生络合作用,形成的络合物会减少药物释放,但胆汁的再摄取会增加进入血液的克拉霉素比例。
-
研究表明食物对药物的释放和片剂的崩解具有显著影响,由于ph和胆酸盐浓度变化,含有标准高脂餐的介质和生理相关介质中的溶出差异显著。
空腹条件下,短暂的胃滞留时间和较长的片剂崩解时间减少了药物的释放,但在进入肠腔后,克拉霉素稳定性改善、片剂崩解快速以及药物溶出均会促进药物的吸收。另一方面,食物摄入后胃内ph升高,克拉霉素稳定性增强,清除率降低,胃滞留时间延长,这些也会延长药物的溶解时间。与空腹相比,进食会降低肝肠首过效应和克拉霉素总清除率,这有助于增大药物的口服生物利用度。然而,小肠中胆汁盐浓度的增加会减慢克拉霉素的溶解速率,使得克拉霉素在餐后条件下不完全吸收。从生理学角度看,克拉霉素在磷酸盐缓冲液、醋酸盐和禁食状态肠液介质中均具有较强的体内外相关性。
gastroplus软件模拟的浓度-时间曲线与体内观测数据吻合良好,该模型具有较好的预测性。小编运用实际pk数据进行模型验证,也表明该模型预测性良好。这些发现表明,体外和pbpk方法可作为预测口服药物食物效应的可靠工具,在开发仿制药过程中,可无需过多的耗时和进行昂贵的临床研究。
-end-


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。


 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 北京新领先
北京新领先 新领先药讯
新领先药讯 010-61006450
010-61006450
