 hotline服务热线:010-61006450
hotline服务热线:010-61006450
汇总 | ind申报流程中美对比(附详细流程图)-九游会电竞
企业需要向美国食品药品监督管理局(fda)提交 ind 申请。一般来说,化药的新药临床申请被称为ind、生产申请被称为nda,仿制药生产申请称为anda,而生物药的申请则被称为bla(biologics license applicatio)。
企业需要向国家药品监督管理局(nmpa)提交申请,申请类型通过受理号加以区分。
-
第一位:c表示国产,j表示进口
-
第二位:x表示新药,y表示已有国家标准(即仿制药)
-
第三位:h表示化学药品,z表示中药,s表示生物制品,f表示辅料
-
第四位:l表示申请临床,s表示申请上市(即生产),b表示补充申请,z表示再注册,f表示分包装,r表示复审
申报详细流程图
美国:
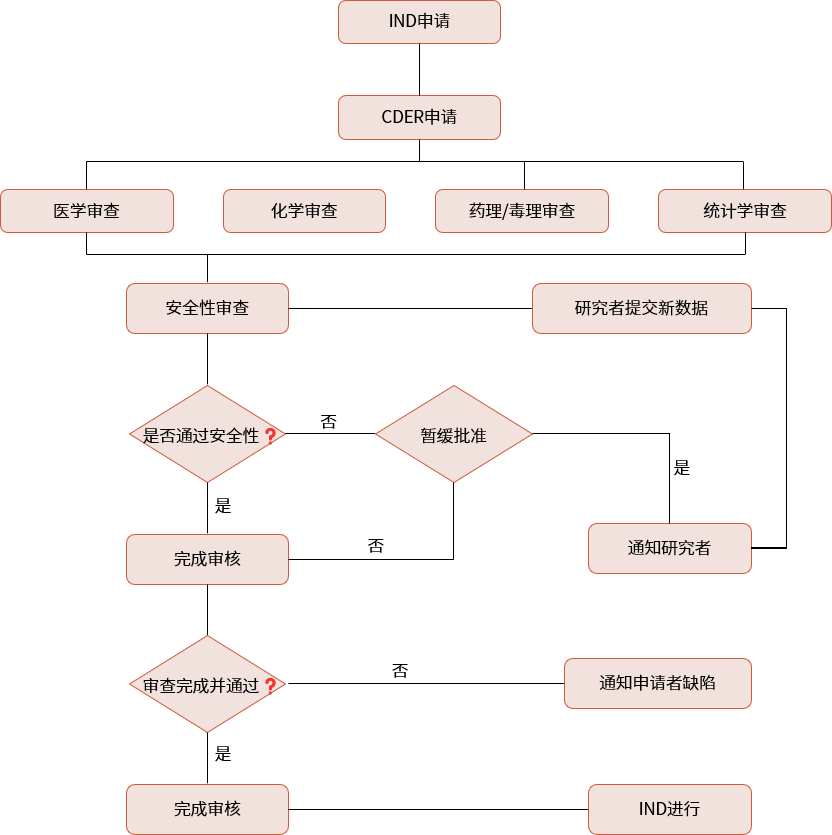
▲ ind申报流程
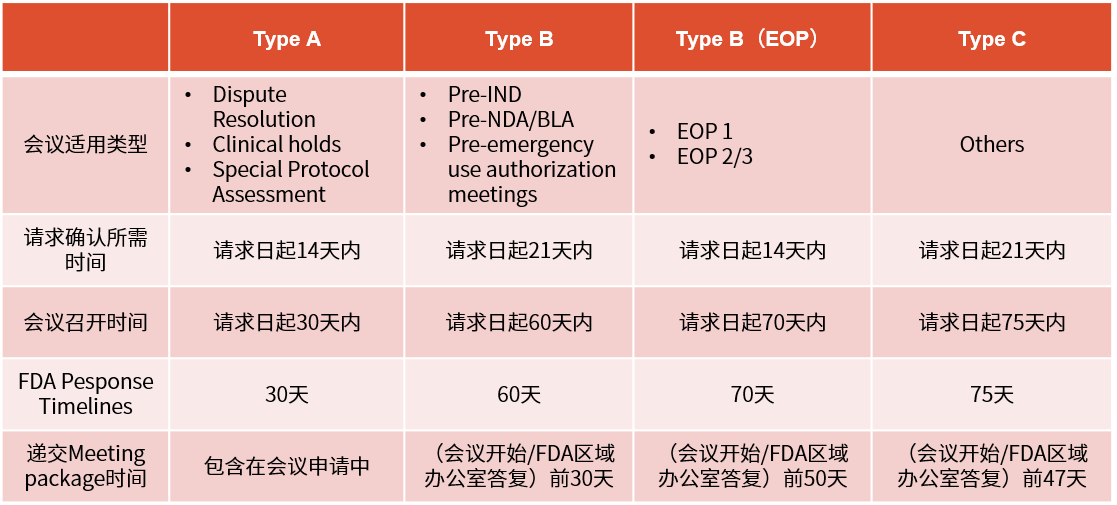
▲ 临床试验申请前会议(pre-ind会议)要求及时限
中国:
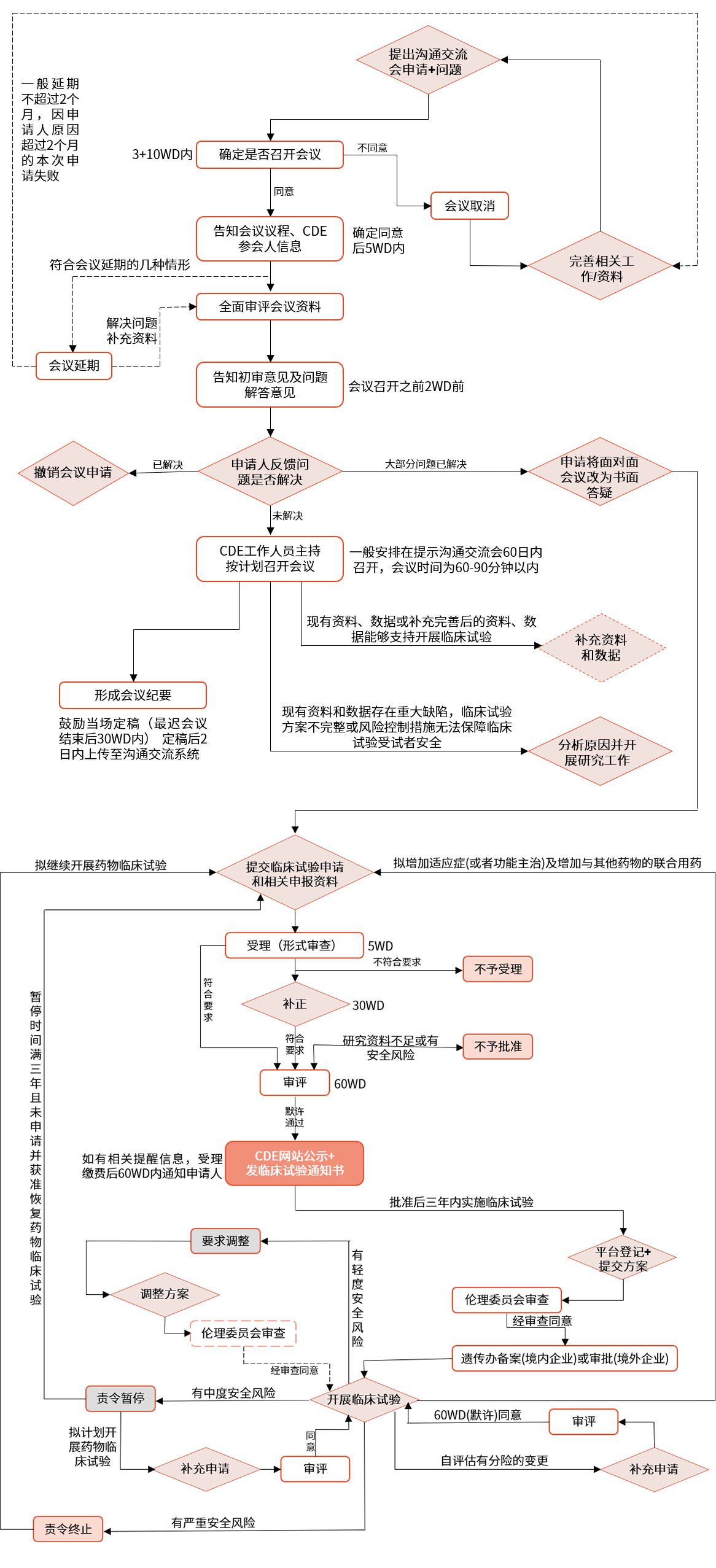
▲ pre-ind沟通交流会、药物临床试验申请流程图
中美申报对比
❖ 整体架构
-
美国:美国fda下设有药品评估和研究中心(cder,center for drug evaluation and research),cder下设多个办公室,审评以治疗领域来划分。
-
中国:nmpa下设药品评审中心(cde,center for drug evaluation),cde下设多个审评处,如化学药审评一处、化学药审评二处、生物制品审评处等。这些审评处分别负责不同类型药品的审评工作。
fda的审评部门是以治疗领域来划分的。nmpa则是根据审评功能划分,包括中药、化学药、生物制品、药理毒理,这是二者在整体构架上的不同。
❖ 沟通交流会
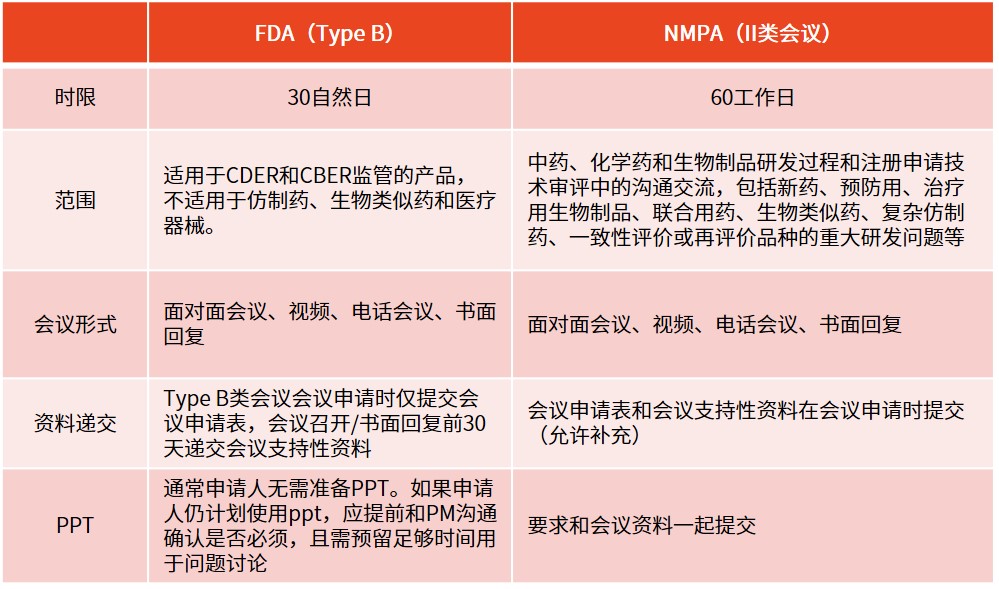
※ pre-ind的重要性(成本、监管机构关系)除了会前准备、会议本身及其后续的沟通成本(包括记录会议内容和回应或澄清机构的问题)外,pre-ind会议不会给申请人带来额外费用。通过避免无谓的研究,并尽早确保与监管机构的策略保持一致,申请人能更快推进产品上市,这相当于减少了项目成本。同时,申请人本身也希望通过减少成本来尽可能减少投资者融资的轮次,因为这通常伴随着各种条件。
最重要的是,pre-ind会议为申请人和监管机构的项目经理提供了一个相互了解、打好合作基础的机会。这样的会议还特别有助于申请人向监管机构介绍那些从未在人体中进行过测试的新分子实体(nme),为双方后续的合作打下了坚实的基础。
❖ 审评过程
-
美国:企业需要在临床试验前向fda提交ind申请。fda有30天的时间对申请进行评估。在30天内,fda会决定是否批准临床试验或者要求额外信息。如果30天内未收到fda的异议,则可视为默许,企业可开始临床试验。对于ind申请,fda不收取审评费用。(与之相对的是上市申请300多万美元的审评费用)
-
中国:企业需要向 nmpa 提交临床试验申请,经过专家评审后,获得批准或默示许可后才能进行临床试验。临床审评收取一定费用。
-end-



转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 北京新领先
北京新领先 新领先药讯
新领先药讯 010-61006450
010-61006450
